Welcome to the Computational Sustainable Research and Drug Design Lab
About Us
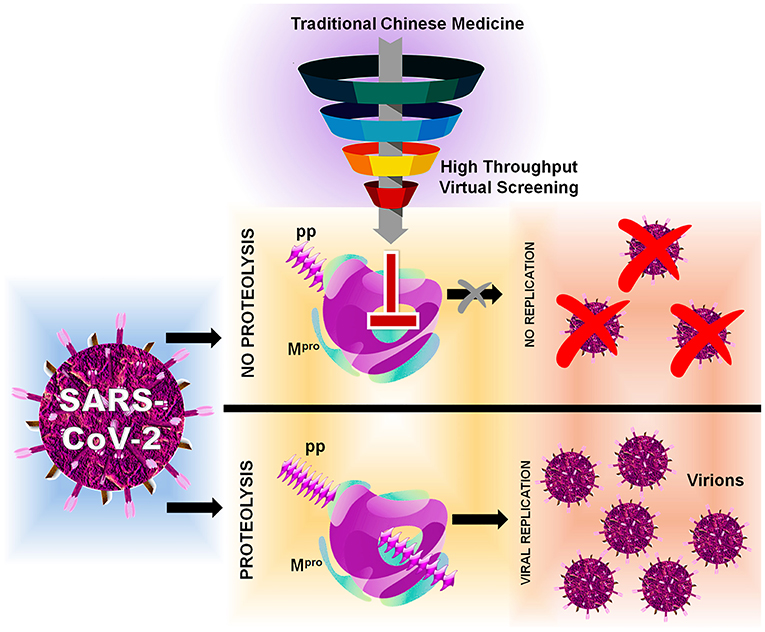
The Computational Sustainable Research and Drug Design Lab (CSRDD) Laboratory integrates advanced computational methodologies with cutting-edge biotechnology to tackle critical challenges in drug discovery and environmental sustainability. We specialize in developing novel therapeutic agents targeting infectious diseases, cancer, and pathogenic organisms through sophisticated molecular modeling, virtual screening, and ligand-based design approaches. Our expanding research portfolio encompasses marine biotechnology and blue carbon economy initiatives, focusing on methanotrophic bacteria in estuarine and mangrove ecosystems to convert CO₂ and methane emissions into sustainable products through enzymatic engineering and mutation studies. we investigate marine-derived bioactive compounds for pharmaceutical applications. Through high-performance computational simulations, QSAR modeling, and rigorous experimental validation, our laboratory bridges theoretical insights with practical solutions, driving innovation in targeted therapeutics and climate change mitigation technologies while developing proprietary software platforms for next-generation biotechnological applications.